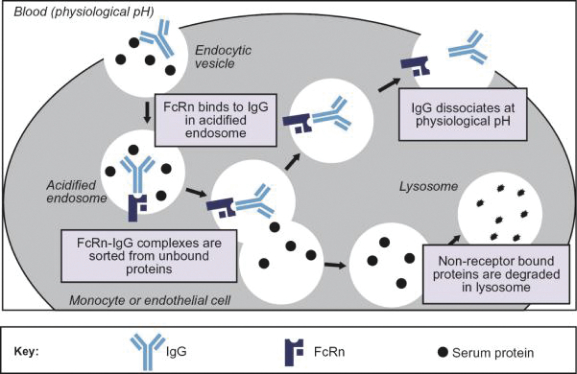
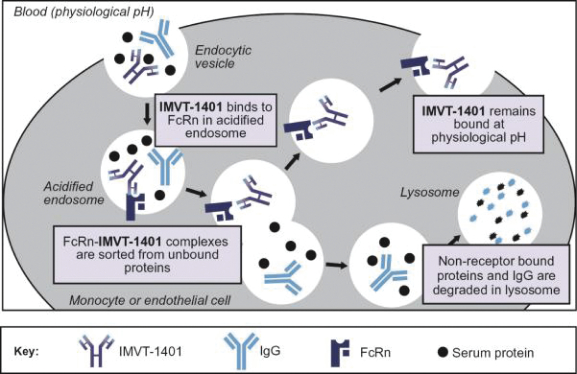
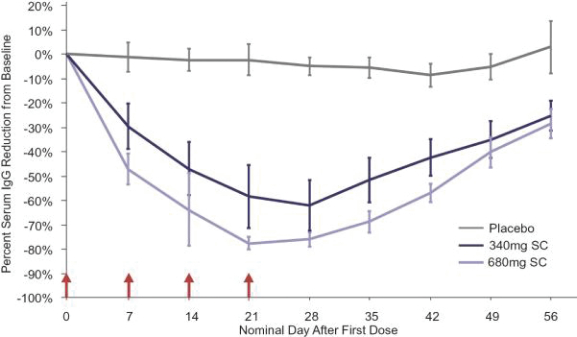
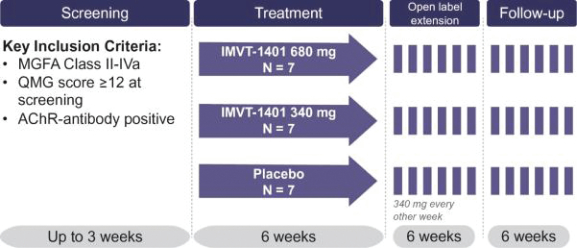
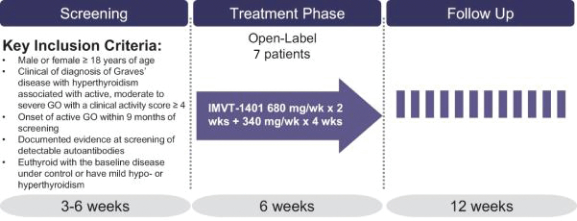
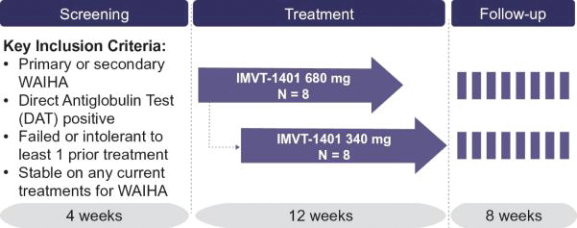
Filed Pursuant to Rule 424(b)(3)
Registration Statement No. 333-235975
PROSPECTUS

17,139,969 Shares of Common Stock
The selling stockholders named in this prospectus or their permitted transferees (the “selling stockholders”) may offer and sell from time to time up to 11,389,969 shares of our common stock, par value $0.0001 per share, consisting of:
| • | up to 5,160,409 shares of common stock issued pursuant to that certain Share Exchange Agreement, dated as of September 29, 2019 (the “Share Exchange Agreement”), by and among Immunovant, Inc. (formerly Health Sciences Acquisitions Corporation (“HSAC”)), a Delaware corporation, Immunovant Sciences Ltd. (“ISL”), a Bermuda exempted limited company, the stockholders of ISL and Roivant Sciences Ltd., a Bermuda exempted limited company (“Roivant” or “RSL”), as representative of such stockholders; |
| • | up to 2,452,062 additional shares of common stock that may be issued pursuant to the Share Exchange Agreement upon the satisfaction of certain conditions, as described under “Certain Relationships and Related Party Transactions—Earnout Payments;” |
| • | up to 2,875,000 shares of common stock held by Health Sciences Holdings, LLC (the “Sponsor”); and |
| • | up to 902,498 shares of common stock purchased by RTW Master Fund Ltd., RTW Innovation Master Fund, Ltd. and RTW Venture Fund Limited (collectively, the “Selling RTW Entities”) in open market transactions. |
In addition, this prospectus relates to the offer and sale of up to 5,750,000 shares of common stock that are issuable by us upon the exercise of outstanding warrants that were previously registered (the “warrants”).
The selling stockholders may offer, sell or distribute all or a portion of the securities hereby registered publicly or through private transactions at prevailing market prices or at negotiated prices. We will not receive any of the proceeds from such sales of the shares of common stock. We will bear all costs, expenses and fees in connection with the registration of these securities, including with regard to compliance with state securities or “blue sky” laws. The selling stockholders will bear all commissions and discounts, if any, attributable to their sale of shares of common stock. See the section titled “Plan of Distribution.”
Our common stock is listed on the Nasdaq Capital Market (“Nasdaq”) under the symbol “IMVT.” On April 9, 2020, the last reported sale price of our common stock on Nasdaq was $15.54 per share.
RSL is currently our majority stockholder, and we are a “controlled company” within the meaning of the listing rules of Nasdaq. In addition, RSL has the right to elect a certain number of Series A Preferred Directors to our board of directors, in accordance with our amended and restated certificate of incorporation. These directors currently hold a majority of the voting power on all matters presented to the board of directors. See “Description of Capital Stock—Series A Preferred Directors.”
We are an “emerging growth company” as defined under the federal securities laws and, as such, have elected to comply with certain reduced public company reporting requirements for this prospectus and our other filings with the Securities and Exchange Commission.
Investing in our securities involves risks. See “Risk Factors” beginning on page 7.
Neither the Securities and Exchange Commission in the United States nor any other regulatory body has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
Prospectus dated April 9, 2020